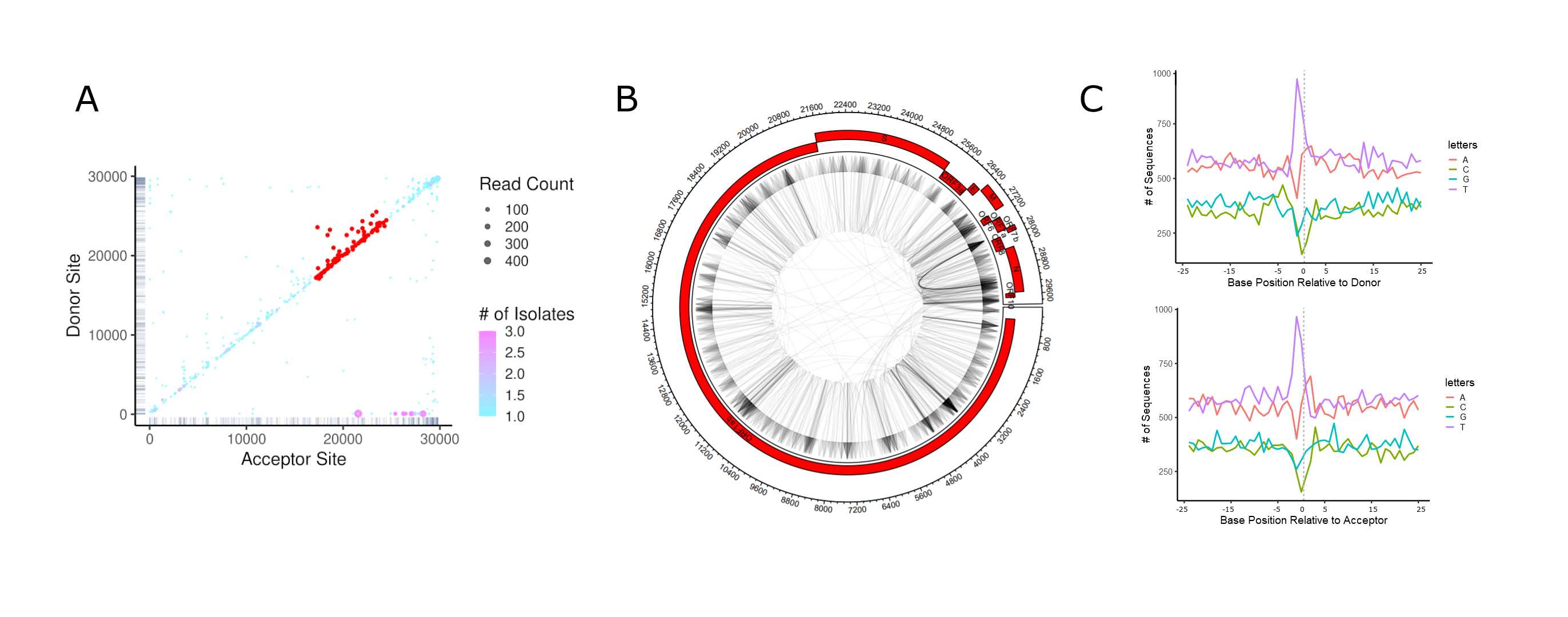
Analyzing viral recombination
Genetic recombination is a tremendous source of intra-host diversity in viruses and is critical for their ability to rapidly adapt to new environments or fitness challenges. While viruses are routinely characterized using high-throughput sequencing techniques, characterizing the genetic products of recombination in next-generation sequencing data remains a challenge.
Viral recombination events can be highly diverse and variable in nature, including simple duplications and deletions, or more complex events such as copy/snap-back recombination, inter-virus or inter-segment recombination and insertions of host nucleic acids. Due to the variable mechanisms driving virus recombination and the different selection pressures acting on the progeny, recombination junctions rarely adhere to simple canonical sites or sequences. Furthermore, numerous different events may be present simultaneously in a viral population, yielding a complex mutational landscape.
What is ViReMa?
ViReMa is an algorithm developed by the Routh and Johnson Labs providing “a versatile platform for rapid, sensitive and nucleotide-resolution detection of recombination junctions in viral genomes using next-generation sequencing data”. Code for ViReMa is written in Python and can be downloaded here.
Read the updated ViReMa paper with vignettes:
Sotcheff S, Zhou Y, Sun Y, Johnson JE, Torbett B, Routh AL. ViReMa: A Virus Recombination Mapper of Next-Generation Sequencing data characterizes diverse recombinant viral nucleic acids. bioRxiv. 2022:2022.03.12.484090. doi:10.1101/2022.03.12.484090
Read the original ViReMa paper:
Andrew Routh, John E. Johnson, Discovery of functional genomic motifs in viruses with ViReMa-a Virus Recombination Mapper-for analysis of next-generation sequencing data, Nucleic Acids Research, Volume 42, Issue 2, 1 January 2014, Page e11, https://doi.org/10.1093/nar/gkt916
Read more articles using ViReMa